Synthese gegen die Stoppuhr
Neuartiges Radiopharmakon zur Diagnostik tumorrelevanter Transportproteine entwickelt
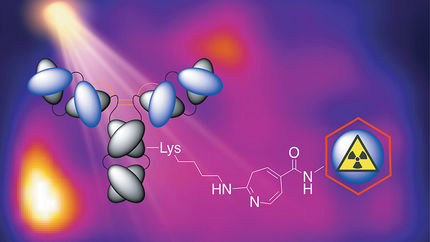
Radioaktiv markierte Moleküle, sogenannte Radiotracer, helfen Nuklearmedizinern beim Aufspüren und zielgenauen Bekämpfen von Tumoren. Deren Entstehung ist oft Folge krankhaft veränderter Stoffwechselprozesse. Ein Team von Wissenschaftlern des Helmholtz-Zentrums Dresden-Rossendorf (HZDR) hat nun erstmalig einen mit dem Fluor-Isotop 18F markierten Radiotracer entwickelt, der spezielle, oft in den Zellmembranen von Krebszellen auftretende Transportproteine mit Hilfe der Positronen-Emissions-Tomographie sichtbar machen kann. Die Forscher haben sich dabei für einen ungewöhnlichen radiochemischen Syntheseansatz entschieden, worüber sie in der Fachzeitschrift Scientific Reports berichten.

Das [18F]FACH-Team (v.l.): Dr. Daniel Gündel, Dr. Masoud Sadeghzadeh, Dr. Magali Toussaint, Dr. Barbara Wenzel, Dr. Winnie Deuther-Conrad, Dr. Rares-Petru Moldovan, Dr. Friedrich-Alexander Ludwig und Prof. Peter Brust.
HZDR/ Bodo Tiedemann
Bösartige Tumore stellen beim Stoffwechsel vermehrt eine bestimmte Art von Transportproteinen bereit. Mit ihnen wird zum Beispiel das Stoffwechsel-Zwischenprodukt Laktat in bestimmte Tumorzellen eingeschleust und gleichzeitig aus anderen abtransportiert – eine Strategie, um die Apoptose, eine Form des programmierten Zelltods, zu verhindern, die bei einem gesunden Stoffwechsel zum Absterben des Tumors führen würde.
„Dieser Zusammenhang wurde bei einer Vielzahl von Tumortypen beobachtet. Deshalb gelten sogenannte Monocarboxylat-Transporter hinsichtlich der Behandlung eines breiten Spektrums verschiedener Krebsarten als Schlüsselproteine, deren Manipulation zum Therapieerfolg führen kann“, erläutert Prof. Peter Brust. Der Leiter der Abteilung Neuroradiopharmaka an der HZDR-Forschungsstelle Leipzig arbeitet an aktuellen Fragestellungen der Radiopharmazie mit dem Schwerpunkt Hirnforschung. „Dazu gehört auch die synthetische Entwicklung moderner Radiotracer, denen im Kampf gegen Krebs und dabei insbesondere aggressive Hirntumoren eine besondere Stellung zukommt“, umreißt Brust die Motivation seines Teams.
In molekularbiologischen und präklinischen Studien hatten Wissenschaftler bereits versucht, die Aktivität der Monocarboxylat-Transporter (MCT) durch den Einsatz von bestimmten kleinen organischen Molekülen mit ausgeprägter Hemmwirkung (etwa das sogenannte α-CHC) zu blockieren. Erste Ergebnisse zeigten, dass die Unterbrechung des Laktatflusses eine sehr effektive therapeutische Strategie gegen das Wachstum von bösartigen Tumoren sein kann.
Radiopharmaka für die nicht-invasive Bildgebung
Neben dem therapeutischen Interesse eröffnet die Rolle der MCT im Stoffwechsel vor allem auch neue diagnostische Möglichkeiten: Sie kommen als wertvolle Biomarker bei vielen Krebserkrankungen in Frage, etwa durch Einsatz der Positronen-Emissions-Tomographie (PET).
Diese Methode verwendet Radionuklide, die positiv geladene Elementarteilchen aussenden, sogenannte Positronen. Dem Patienten wird zunächst ein Radiopharmakon verabreicht, ein Molekül mit einem angekoppelten radioaktiven Atom wie etwa 18F, das Positronen abstrahlt. Bei der Wechselwirkung eines Positrons mit einem Elektron im Körper wird Strahlung in Form zweier hochenergetischer Photonen in diametral entgegengesetzte Richtungen ausgesandt, die die ringförmig um den Patienten angeordneten Detektoren aufzeichnen. Aus diesem Abbild der Stoffwechselprozesse können die Ärzte auf die räumliche Verteilung des Radiopharmakons im Körperinneren und somit auf krankhafte Veränderungen schließen.
Ziel: Schnelle, automatisierte Radiopharmakon-Synthese für den klinischen Alltag
Trotz dieses Potentials als therapeutische Zielstruktur im Kampf gegen Krebs wurden in den vergangenen Jahren kaum radioaktiv markierte MCT-Hemmer auf ihre Eignung in diagnostischen bildgebenden Verfahren wie der PET erforscht. „Wir haben nun einen Strukturverwandten des bekannten MCT-Hemmers α-CHC entwickelt und in einem aufwendigen Verfahren erfolgreich mit dem PET-Radionuklid 18F gekoppelt. Dessen relativ kurze Halbwertszeit von 110 Minuten gewährleistet eine für den Patienten tolerable Strahlenbelastung“, beschreibt der Koordinator der Versuche, Dr. Masoud Sadeghzadeh, das Herangehen der Leipziger Chemiker.
Nach ersten vielversprechenden präklinischen Untersuchungen ihrer neuen Verbindung [18F]FACH überarbeiteten die Wissenschaftler ihren Syntheseweg. „Die Herausforderung dabei ist, dass wir den Radiotracer schnell genug herstellen, um die strahlenden Eigenschaften von 18F überhaupt praktisch nutzen zu können“, fasst die Radiochemikerin Dr. Barbara Wenzel zusammen. Denn die Zeitspanne der Nutzbarkeit wird von der Halbwertszeit des Radionuklids vorgegeben. Während die Chemiker manuell zunächst noch 160 Minuten für die Herstellung des neuen Radiotracers benötigten, konnten sie diese Zeit in einem modifizierten Ansatz halbieren.
„Der Clou unserer Synthese ist, dass sie ohne den Einbau einer Schutzgruppe auskommt. Dieser nun hinfällige Zwischenschritt ist üblicherweise nötig, um reaktive Teile eines Moleküls vor unbeabsichtigten Nebenreaktionen zu bewahren“, fügt Barbara Wenzel hinzu. Dadurch haben die Wissenschaftler das Verfahren erheblich vereinfacht und es für die Überführung in ein automatisiertes Synthesemodul angepasst – unabdingbare Voraussetzung für die nun geplanten Tumoruntersuchungen und eine mögliche zukünftige nuklearmedizinische Verwendung.
Originalveröffentlichung
M. Sadeghzadeh, R.-P. Moldovan, R. Teodoro, P. Brust, B. Wenzel; "One-step radiosynthesis of the MCTs imaging agent [18F]FACH by aliphatic 18F-labelling of a methylsulfonate precursor containing an unprotected carboxylic acid group"; Scientific Reports; 2019.
M. Sadeghzadeh, R.-P. Moldovan, S. Fischer, B. Wenzel, F.-A. Ludwig, R. Teodoro, W. Deuther-Conrad, S. Jonnalagadda, S. K. Jonnalagadda, E. Gudelis, A. Šačkus, K. Higuchi, V. Ganapathy, V. R. Mereddy, L. R. Drewes, P. Brust; "Development and radiosynthesis of the first 18F-labeled inhibitor of monocarboxylate transporters (MCTs)"; Journal of Labelled Compounds and Radiopharmaceuticals; 2019.
Weitere News aus dem Ressort Wissenschaft