Neues Tool integriert Mikrobiom- und genetische Wirtssequenzierungsanalyse
Neue Software erleichtert die Untersuchung der Beziehungen zwischen einem Wirt, seinem Mikrobiom und Krankheitserregern wie HIV oder SARS-CoV-2
Forscher des Texas Biomedical Research Institute und der Tulane University haben ein neues Software-Tool entwickelt, mit dem sich genetische Informationen über einen Wirt und sein Mikrobiom einfacher, schneller und kostengünstiger gleichzeitig analysieren lassen. Die Software mit der Bezeichnung „Meta-Transkriptom-Detektor“ (MTD) kann von einer Vielzahl von Mikrobiologen und Arzneimittelentwicklern eingesetzt werden, darunter auch von Forschern, die sich mit Krankheiten wie bestimmten Krebsarten, COVID-19, HIV/AIDS, Malaria und vielen anderen mit Mikroorganismen zusammenhängenden Gesundheitszuständen befassen. Das Tool wurde kürzlich in der Zeitschrift Briefings in Bioinformatics veröffentlicht.
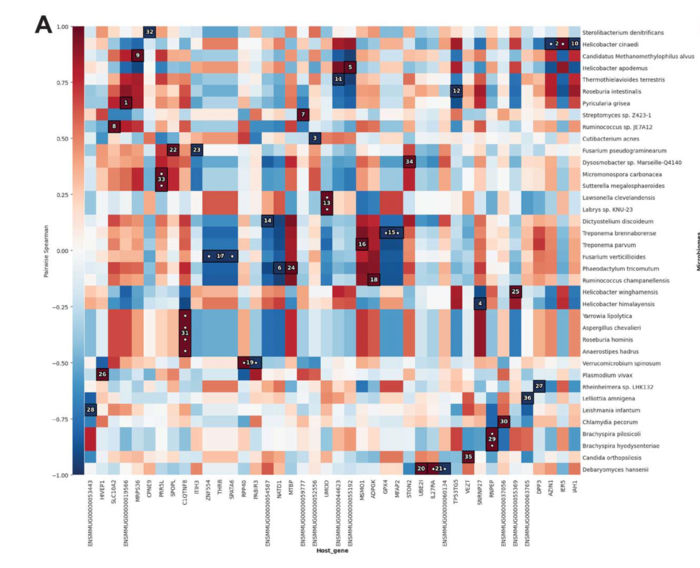
MTD analysiert die Aktivität der Genexpression im Wirt und im Mikrobiom gleichzeitig und erstellt automatisch eine Grafik, die die Korrelationen zwischen den beiden darstellt. Ein dunkleres Rot zeigt eine stärkere positive Korrelation an, was bedeutet, dass die Hoch- oder Herunterregulierung eines bestimmten Wirtsgens oder -wegs mit der Hoch- oder Herunterregulierung eines bestimmten Mikrobentyps in derselben Richtung verbunden ist. Dunkleres Blau zeigt eine stärkere negative Korrelation an, was bedeutet, dass ein Wirtsgen oder -weg weniger aktiv ist, die Mikrobenart jedoch aktiver ist. Die Analyse zeigt zwar nicht, dass das eine das andere verursacht, aber sie kann Zusammenhänge aufzeigen, die Forscher weiter untersuchen sollten, um die Ursache-Wirkungs-Beziehung für die mögliche Entwicklung von Behandlungen zu verstehen.
Texas Biomed
„Es ist sehr benutzerfreundlich, vor allem für Forscher mit wenig bis gar keinen Kenntnissen in Bioinformatik“, sagt Associate Professor Binhua „Julie“ Ling, PhD, die das Host-Pathogen Interactions Research Program am Texas Biomed mit leitet und die Hauptautorin der Veröffentlichung ist. „Man muss nur eine Zeile Code schreiben, um bestimmte Parameter einzustellen, und die Software erledigt den Rest automatisch.“
MTD ermöglicht es den Forschern, einen umfassenden Überblick über die im Wirt vorhandenen Mikroben zu erhalten – einschließlich der zahlreichen „guten Bakterien“, die normalerweise auf und in Menschen und Tieren leben, aber auch der schädlichen, wie z.B. Viren, die schwere Krankheiten verursachen. Noch wichtiger ist, dass MTD die Aktivität der Genexpression analysiert, d.h. im Wesentlichen, welche Gene ein- oder ausgeschaltet sind, und zwar sowohl bei den Mikroben als auch beim Wirt, so dass die Forscher leicht Beziehungen zwischen ihnen erkennen können. Wenn man zum Beispiel sieht, welche Gene in einer Mikrobe und im Wirt aktiv sind, könnte das darauf hindeuten, dass die Aktivität der einen Mikrobe von der anderen beeinflusst wird, und das könnte ein mögliches Ziel für eine spätere Behandlung sein, erklärt Ling.

„Wir können in diesem Stadium nicht sagen, ob es sich um Ursache und Wirkung handelt, aber wir können diese Analyse nutzen, um herauszufinden, welche Gene oder Signalwege wir untersuchen sollten – vielleicht solche, die wir vorher nie als zusammenhängend betrachtet haben“, sagt Ling. „MTD kann dazu beitragen, diesen Prozess zu beschleunigen und möglicherweise neue Wege in der Forschung und Arzneimittelentwicklung zu eröffnen.
MTD verknüpft mehrere bestehende Softwarepakete und greift auf internationale Datenbanken zurück, die RNA-Sequenzen von mehr als 100.000 Bakterien-, Virus-, Pilz-, Archaeen- und Protozoenarten sowie Sequenzen von Vektoren und Plasmiden enthalten. Die Benutzer können die Datenbank auch mit bestimmten Sequenzen, die sie interessieren, aktualisieren.
Erstautor Fei Wu, PhD, untersuchte zusammen mit Ling, wie sich das Mikrobiom bei jüngeren und älteren Affen mit dem Affen-Immundefizienz-Virus (simian immunodeficiency virus, SIV), der Affenversion von HIV, mit dem Alter verändert.
„Wir mussten die Genexpression des Wirts in einem Arbeitsgang und die Genexpression des Mikrobioms in einem anderen Arbeitsgang analysieren“, sagt Wu. „Wir fragten uns, warum können wir nicht beides gleichzeitig machen?“
Da ihr Labor während der COVID-19-Pandemie umzog, hatten sie die Zeit, sich auf die computergestützte Arbeit zu konzentrieren, und machten sich daran, dieses Problem zu lösen. Wu arbeitete gemeinsam mit Ling und dem Kooperationspartner Yao-Zhong Liu, PhD, außerordentlicher Professor an der Tulane University School of Public Health and Tropical Medicine, an der Entwicklung und Testung der neuen Software.
„Normalerweise verwenden wir Bioinformatiksoftware, um unsere Daten zu analysieren, wir erstellen sie nicht“, sagt Wu. „Es war eine Herausforderung, aber auch aufregend, einen neuen Weg einzuschlagen und jetzt ein Werkzeug zu haben, das nicht nur uns hilft, sondern auch allen anderen Forschern, die RNA-Sequenzierung von Wirten und Mikrobiomen durchführen – von Menschen und Affen bis hin zu Malariaparasiten übertragenden Moskitos und Schistosomenparasiten übertragenden Schnecken.“
Hinweis: Dieser Artikel wurde mit einem Computersystem ohne menschlichen Eingriff übersetzt. LUMITOS bietet diese automatischen Übersetzungen an, um eine größere Bandbreite an aktuellen Nachrichten zu präsentieren. Da dieser Artikel mit automatischer Übersetzung übersetzt wurde, ist es möglich, dass er Fehler im Vokabular, in der Syntax oder in der Grammatik enthält. Den ursprünglichen Artikel in Englisch finden Sie hier.
Originalveröffentlichung

Weitere News aus dem Ressort Wissenschaft




















Diese Produkte könnten Sie interessieren

Limsophy LIMS von AAC Infotray
Optimieren Sie Ihre Laborprozesse mit Limsophy LIMS
Nahtlose Integration und Prozessoptimierung in der Labordatenverwaltung

OMNIS von Metrohm
OMNIS – die Plattform zur Integration der Metrohm Titrando Gerätegeneration
OMNIS ermöglicht die Kombination von Bestandskomponenten mit neuester OMNIS Hard- und Software

LAUDA.LIVE von LAUDA
LAUDA.LIVE - Die digitale Plattform für Ihre Geräteverwaltung
Viefältige Flottenmanagementoptionen für jedes LAUDA Gerät - mit und ohne IoT-Anbindung

Meistgelesene News





Weitere News von unseren anderen Portalen






























Zuletzt betrachtete Inhalte
Max-Planck-Innovation lizenziert Nanoskopieverfahren an Leica Microsystems - Exklusivlizenz für neue Generation optischer Mikroskope
