Kraftmesser für molekulare Bindungen
Haftkraft zwischen einzelnen Molekülen erstmals direkt experimentell ermittelt
Wie stark Superkleber, Haftmagnete und Klemmverbindungen zusammenhalten, lässt sich durch mechanische Belastungstests ziemlich präzise bestimmen. Anders sieht es auf mikroskopischer Ebene aus: Wie stark ein einzelnes Molekül an einer Oberfläche haftet, ließ sich bisher nicht direkt messen. Jülicher Physiker haben in Physical Review Letters eine neue Methode vorgestellt, die dies ändern könnte. Mit einem Rasterkraftmikroskop haben sie erstmals auf direktem Weg die Haftkraft einzelner Moleküle an Oberflächen ermittelt. Dabei konnten sie zum ersten Mal auch gezielt die Beiträge einzelner Bindungstypen wie der Van-der-Waals-Kräfte oder der chemischen Bindungen feststellen.

An einer Quarzstimmgabel (qPlus-Sensor) befestigter Tastkopf des Rasterkraftmikroskops, der von Prof. Joachim Mayer, RWTH Aachen, mit einem „fokussierten Ionenstrahl“ bis aufs einzelne Atom genau zugespitzt wurde, um damit einzelne Moleküle kontrolliert ansteuern zu können.
Forschungszentrum Jülich
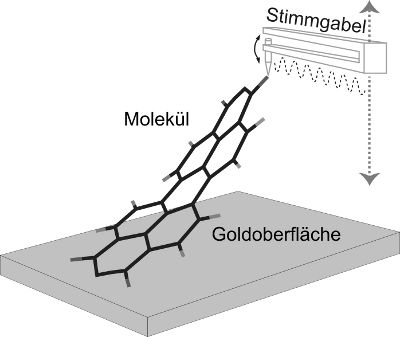
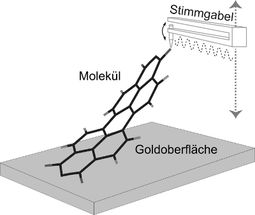
Versuchsskizze: Die auf ein einziges Atom zulaufende Spitze des Rasterkraftmikroskops dockt von oben an einem Sauerstoffatom des zu untersuchenden PTCDA-Moleküls an und hebt es von der Gold-Oberfläche ab. Über Frequenzänderungen von bis zu 50 Hz an der mit 30 kHz schwingenden Mikroskopspitze lassen sich die Bindungskräfte zwischen Molekül und Oberfläche ableiten.
Forschungszentrum Jülich
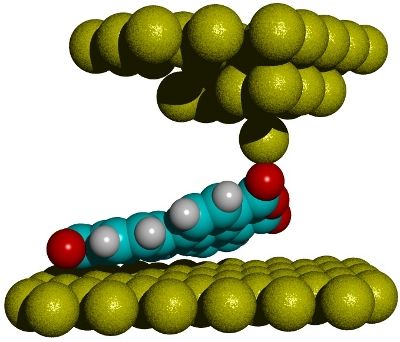
Zur Ermittlung einzelner Werte für verschiedene Bindungstypen werden die Messergebnisse mit Daten aus Computersimulationen verglichen.
Forschungszentrum Jülich

Bisherige experimentelle Methoden lieferten nur eine grobe Vorstellung davon, wie stark einzelne organische Moleküle durch verschiedene, sich gegenseitig überlagernde Kräfte an einer Oberfläche haften. Auch auf theoretischem Weg ist diese Frage nicht eindeutig zu lösen. Die Van-der-Waals-Kräfte etwa entstehen, weil die Elektronen, die sich in einer Art "Elektronenwolke" um das Molekül herum befinden, nicht gleich verteilt sind und diese Verteilung sich zudem ständig ändert. Die ungleichen Ladungsverteilungen beeinflussen sich gegenseitig, was den Effekt noch verstärkt und dazu führt, dass sich die Elektronen noch mehr verschieben. Aufgrund der vielen beteiligten Teilchen und der komplexen Wechselwirkungen lassen sich die resultierenden Kräfte nur annäherungsweise berechnen, mit Methoden, die sich derzeit noch in der Entwicklung befinden. Die neue Jülicher Methode ermöglicht es dagegen erstmalig, den Anteil der Van-der-Waals-Kraft und anderer Bindungstypen präzise experimentell zu bestimmen.
Die Wissenschaftler aus der Helmholtz-Nachwuchsgruppe "Complex Transport Regimes in Low Temperature Scanning Tunneling Microscopy" nutzen für ihr Verfahren unter anderem aus, dass sich die verschiedenen Bindungstypen unterschiedlich stark abschwächen. "Entfernt man ein Molekül kontinuierlich von der Oberfläche, so spürt es anfänglich noch alle Wechselwirkungen, aber schon nach einigen Nanometern bleibt nur die Van der Waals-Anziehung übrig", erläutert Dr. Christian Wagner aus dem Jülicher Peter Grünberg Institut. "Für eine chemische Bindung müssen sich die Elektronenwolken von Molekül und Oberfläche überlappen. Für die Van-der-Waals-Wechselwirkung ist dies nicht notwendig."
"Dieses Verfahren ist eine fundamentale, messtechnische Neuerung in der Oberflächenphysik. Durch seinen universellen Charakter ist es vorstellbar, auch Daten zu gewinnen, die in ganz unterschiedlichen anderen Gebieten, etwa in der Pharma- und Energieforschung, relevant sind. Es ist meines Wissens nach die einzige Möglichkeit, die Bindungsenergie einzelner Moleküle an Oberflächen direkt zu bestimmen", betont der Leiter der Forschungsgruppe, Dr. Ruslan Temirov, vom Jülicher Peter Grünberg Institut.
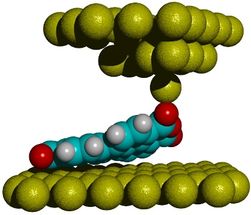
Als Modellmolekül haben die Jülicher Wissenschaftler mit der Verbindung Perylentetracarbonsäuredianhydrid (PTCDA) experimentiert, die aus sieben Kohlenstoffringen und insgesamt sechs Sauerstoffatomen an den Enden besteht. Den Messungen zufolge beträgt die Bindungsenergie eines solchen Einzelmoleküls an eine Gold-Oberfläche rund 2,5 Elektronenvolt (eV) und nicht 2,0 eV, wie durch frühere Berechnungen vorhergesagt. Ein Großteil davon, in der Größenordnung von 100 meV für jedes der 24 Kohlenstoff- und sechs Sauerstoffatome, geht auf Van-der-Waals-Kräfte zurück. Vergleiche sind schwierig. "Die gesamte Bindungsenergie entspricht umgerechnet 4 mal 10 hoch minus 19 Joule – das ist eine Null mit achtzehn Nullen hinter dem Komma. Diese Energie reicht gerade aus, um ein Staubkorn um den Durchmesser eines Wasserstoffatoms anzuheben", so Wagner.
Für ihre Messung platzierten die Forscher das PTCDA-Molekül auf einer Goldunterlage und kontaktierten es an einer Ecke mit dem auf ein einzelnes Atom zugespitzten Tastkopf eines Rasterkraftmikroskops. Dann hoben sie das längliche Molekül schrittweise an, bis es sich vollständig ablöste. Die Bindungskräfte zwischen Molekül und Oberfläche machen sich dabei durch Frequenzänderungen an der schnell schwingenden Mikroskopspitze bemerkbar, die an einer kleinen Quarzstimmgabel klebt.
Um die gesuchte Stärke der verschiedenen Bindungstypen zu ermitteln, haben die Jülicher Physiker die Messergebnisse mit Daten aus Computersimulationen verglichen. In Zusammenarbeit mit Experten vom Jülich Supercomputing Centre (JSC) ermittelten sie präzise, mit welcher Form der Bindung zwischen Molekül und Oberfläche die Experimente am besten übereinstimmen. Dafür wurden insgesamt 100 Millionen unterschiedliche Bindungspotenziale auf dem Jülicher Superrechner JUDGE getestet, für rund 4000 von ihnen wurde der Ablösevorgang komplett simuliert. Die in der Kombination aus Simulation und Experiment gewonnenen Parameter lassen sich ohne weiteren Aufwand auf ähnliche Fälle übertragen. Die Jülicher Physiker konnten damit auch die Bindungskräfte von NTCDA, einem Modellmolekül, das ähnlich wie PTCDA aufgebaut ist, korrekt vorhersagen.
Originalveröffentlichung

Weitere News aus dem Ressort Wissenschaft