Krebsstammzellen nachverfolgen
Was passiert innerhalb und zwischen einzelnen Zellen in den allerersten Stadien der Tumorentwicklung?
Mit Hilfe von Einzelzell-Sequenziertechnologien und einem Mausmodell konnten Forscher die zelluläre Vielfalt ganzer Speicheldrüsentumore umfassend kartieren und den Weg der Krebsstammzellen zurückverfolgen.
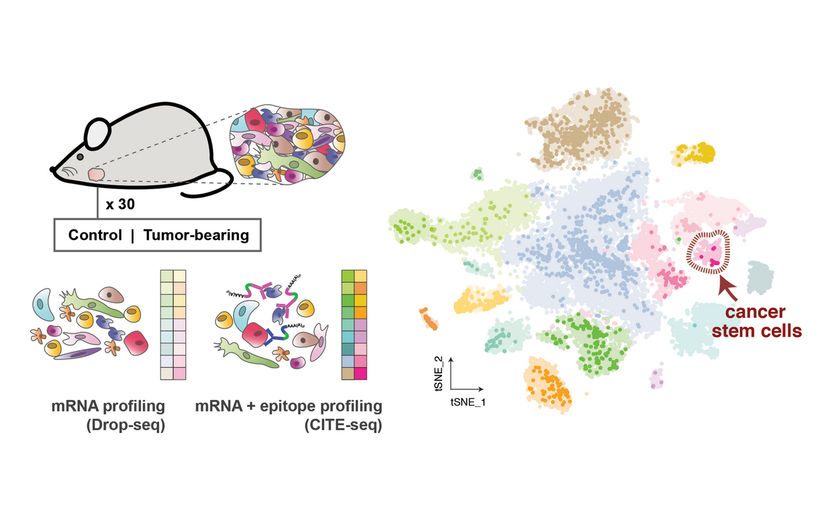
Links: Die Forscher sequenzierten mehr als 26'000 einzelne Zellen von gesunden Mäusen und Mäusen mit Speicheldrüsentumoren. Mit Hilfe verschiedener Strategien analysierten sie sowohl die RNA im Inneren der Zellen (mRNA-Profiling) als auch die Proteine auf der Oberfläche der Zellen (Epitop-Profiling), um die Zellen zu identifizieren. Rechts: Das Ergebnis war ein umfassender Zellatlas der gesunden und tumortragenden Speicheldrüse (hier gezeigt). Er zeigte unterschiedliche Populationen der verschiedenen Zelltypen, darunter eine sehr kleine Gruppe von Krebsstammzellen.
© Samantha Praktiknjo, MDC
Zwei Forschungsteams des Max-Delbrück-Centrums für Molekulare Medizin in der Helmholtz-Gemeinschaft (MDC) und ihre Kooperationspartner haben einen detaillierten Zellatlas eines ganzen Speicheldrüsentumors in einem Mausmodell erstellt. Er bildet einzelne Zellen im gesamten Tumor und dem umgebenden Gewebe ab. Dieser „Einzelzell“-Ansatz hat wichtige Erkenntnisse über Veränderungen der zellulären Zusammensetzung in den frühesten Stadien der Krebsentstehung geliefert und wurde gerade in „Nature Communications“ veröffentlicht.
Ein solider Tumor ist nicht, wie viele vielleicht annehmen, ein Klumpen von Zellen, die alle gleich sind. Vielmehr handelt es sich um eine Mischung aus vielen verschiedenen Zelltypen, darunter neben den eigentlichen Tumorzellen auch eine Vielzahl von Stroma- und Immunzellen.

„Herkömmliche Methoden in der Molekularbiologie betrachten eine Probe oft als Ganzes. Das erfasst aber die Komplexität dieser Probe nicht", sagte Dr. Samantha Praktiknjo, Wissenschaftlerin und Erstautorin vom MDC-Labor für „Systembiologie der genregulatorischen Elemente“ unter der Leitung von Professor Nikolaus Rajewsky am Berliner Institut für Medizinische Systembiologie (BIMSB). Die verschiedenen Zellen innerhalb eines Tumors und ihr Zusammenspiel im Detail zu verstehen könnte helfen, effektivere Behandlungsstrategien zu identifizieren.
Die Macht der Datenfülle
Das Team nutzte die in der AG Rajewsky entwickelten Technologien zur Einzelzell-RNA-Sequenzierung und eine neue Epitop-Profiling-Methode, um den Zellatlas zu erstellen. Es identifizierte die Zellen, die spezifisch für den Tumor waren, indem es die Reproduzierbarkeit und den großen Umfang ihrer Daten optimal ausschöpfte.
Möglich war das dank eines Mausmodells, das im MDC-Labor für „Signalvermittlung in Entwicklung und Krebsentstehung“ unter der Leitung von Professor Walter Birchmeier entwickelt wurde. Das Erbgut dieser Mäuse enthält Mutationen, die ein Plattenepithelkarzinom der Speicheldrüse auslösen. So stehen kontinuierlich genetisch ähnliche Tumore für die Forschung bereit, die schon in den frühesten Entwicklungsstadien sequenziert werden können. Bei menschlichen Patienten wäre das nahezu unmöglich.
„Bei einem Patienten oder einer Patientin ist der Tumor bereits entwickelt. Man kann nicht einfach die Zeit zurückspulen und sich ansehen, wie der Krebs begonnen hat“, sagte Dr. Benedikt Obermayer, einer der Erstautoren, der jetzt am Berlin Institute for Health (BIH) arbeitet. „Wir haben hier ein kontrolliertes Modell, bei dem wir zusehen können, was passiert.“ Und Dr. Qionghua Zhu, die dritte Erstautorin und ehemalige Doktorandin in der AG Birchmeier, fügte hinzu: „Um Krebs wirksam zu bekämpfen, müssen wir die Treibermutationen finden. Diese Methode gibt uns Anhaltspunkte über die Evolution eines Tumors.“
Die Sequenziertechnologien sind inzwischen so weit fortgeschritten, dass sowohl die RNA innerhalb einzelner Zellen, eine nach der anderen, als auch die Proteine auf den Zelloberflächen im Gewebe schnell und kostengünstig sequenziert werden können. Während andere Methoden das Gewebe zerkleinern und feststellen, welche Gene und Moleküle in der Mischung vorhanden sind, wird beim Einzelzellansatz genau bestimmt, wie viel von jedem Zelltyp vorhanden ist und welche Gene und Moleküle mit welcher Zelle assoziiert sind.
Für die aktuelle Studie sequenzierten die Forscher mehr als 26.000 einzelne Speicheldrüsenzellen von Mäusen mit Tumoren und von gesunden Mäusen. Mit Hilfe von Computermodellen analysierten sie die riesige Datenmenge und identifizierten jede einzelne Zelle und sortierten sie in Gruppen - wie zum Beispiel Stromazellen, Immunzellen, speichelproduzierende Zellen, Krebszellen - basierend auf den Hunderten von exprimierten Genen und den vorhandenen Molekülen.
Eine Überraschung
Der Einzelzellansatz offenbarte den Forschern eine Überraschung: „Als ich die Daten sah, dachte ich: Wo ist der Tumor?“, sagte Obermayer. Die Population der Krebsstammzellen im Tumor war extrem klein – weniger als ein Prozent aller charakterisierten Zellen im Gewebe. Weil sie so selten sind, hängt die Untersuchung dieser Zellen sonst noch sehr von Annahmen über Oberflächenmarker ab und wird oft in Zellkulturen durchgeführt. In diesem Fall konnten die Autor*innen jedoch die Krebsstammzellen mit ihrer Einzelzellanalyse direkt in den Proben des soliden Tumors nachweisen.
Darüber hinaus konnte das Team die Entwicklung der verschiedenen Zelltypen während verschiedener Stadien der Tumorentwicklung vorhersagen. Ihr Modell legt nahe, dass die Krebsstammzellen aus krebsartigen Basalzellen entstehen, sich dann in einen weiteren Subtyp entwickeln, bevor sie schließlich zu einer Population von Zellen werden, die den Luminalzellen ähnlich sind. Dieser Zelltyp kommt in normalen, gesunden Speicheldrüsen vor.
Dieser Verlauf unterstützt die folgende These: Wenn in den Basalzellen dieses soliden Tumormodells etwas schief läuft, werden diese Zellen dazu veranlasst, sich in Krebsstammzellen zu verwandeln, die dann wiederum zu einem anderen Zelltyp werden können. „Was ich faszinierend fand, war die klare Abfolge der Signale und Ereignisse, die von Vorläufern zu den Nachkommen der Krebsstammzellen übergehen“, sagte Praktiknjo.
Nächste Schritte
Um zu verifizieren, dass einzelne Zellen all diese Phasen durchlaufen, ist noch weitere Forschung nötig. Das gilt auch für das zelluläre und molekulare Zusammenspiel, die das Tumorwachstum antreiben. Das Team geht davon aus, dass ihr Ansatz auch auf andere Krebsarten anwendbar ist.
„Für mich besteht die wichtigste konzeptionelle Erkenntnis darin, dass wir Ideen aus der einzelzellbasierten Entwicklungsbiologie anwenden können, um das molekulare Fortschreiten der Tumorentstehung zu rekonstruieren“, sagte Professor Nikolaus Rajewsky, der das MDC-Labor für „Systembiologie von genregulatorischen Elementen“ leitet und wissenschaftlicher Direktor des BIMSB ist.
Originalveröffentlichung
Weitere News aus dem Ressort Wissenschaft



















Meistgelesene News






Weitere News von unseren anderen Portalen






























Verwandte Inhalte finden Sie in den Themenwelten
Themenwelt Zellanalyse
Die Zellanalyse ermöglicht es uns, Zellen in ihren vielfältigen Facetten zu erforschen und zu verstehen. Von der Einzelzellanalyse über die Durchflusszytometrie bis hin zur Bildgebungstechnologie – die Zellanalyse bietet uns wertvolle Einblicke in die Struktur, Funktion und Interaktion von Zellen. Ob in der Medizin, der biologischen Forschung oder der Pharmakologie – die Zellanalyse revolutioniert unser Verständnis von Krankheiten, Entwicklung und Behandlungsmöglichkeiten.
Themenwelt Zellanalyse
Die Zellanalyse ermöglicht es uns, Zellen in ihren vielfältigen Facetten zu erforschen und zu verstehen. Von der Einzelzellanalyse über die Durchflusszytometrie bis hin zur Bildgebungstechnologie – die Zellanalyse bietet uns wertvolle Einblicke in die Struktur, Funktion und Interaktion von Zellen. Ob in der Medizin, der biologischen Forschung oder der Pharmakologie – die Zellanalyse revolutioniert unser Verständnis von Krankheiten, Entwicklung und Behandlungsmöglichkeiten.