Pflanzenerbgut mit hoher Auflösung entpuzzeln
Neues Softwaretool ermöglicht die Zuordnung zu den richtigen Kopien – das „Phasing“ – mit hoher Genauigkeit
Die Aufschlüsselung insbesondere eines pflanzlichen Genoms ist sehr aufwändig und fehlerträchtig. Grund ist, dass alle Chromosomen in mehreren, sehr ähnlichen Kopien vorliegen. Ein Forschungsteam von Bioinformatikern der Heinrich-Heine-Universität Düsseldorf (HHU) hat nun ein Softwaretool entwickelt, mit dem die Zuordnung zu den richtigen Kopien – das „Phasing“ – mit hoher Genauigkeit möglich ist.

Symbolbild
Capri23auto, pixabay.com

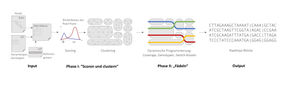
Funktionsprinzip des bioinformatischen Softwaretools „WhatsHap polyphase“.
Gunnar Klau/HHU

Das Erbgut aller höheren Lebewesen ist im Zellkern auf Chromosomen gespeichert. Diese bestehen aus Strängen des Moleküls DNA. Die Erbinformation selbst ist in einer Abfolge von hintereinanderliegenden Basenpaaren kodiert, wobei es vier „Buchstaben“ gibt, die durch die Moleküle Adenin (A), Cytosin (C), Guanin (G) und Tyrosin (T) repräsentiert sind.
Verschiedene Lebewesen haben unterschiedliche Zahlen von Chromosomen: beim Menschen sind es 23 unterschiedliche, bei der Kartoffel 12, beim Weizen 7. Zusätzlich gibt es unterschiedliche Kopien oder „Haplotypen“ der Chromosomen: Beim Menschen liegen zwei Kopien vor – eine kommt von der Mutter, eine vom Vater –, bei Kartoffeln sind es vier, bei Weizen sogar sechs. Lebewesen mit zwei Kopien nennt mit „diploid“, solche mit einer größeren Zahl „polyploid“. Die Kopien sind fast identisch, aber eben nicht ganz; die Unterschiede machen die Variabilität der Organismen innerhalb einer Population aus.

Um die Erbinformation zu entschlüsseln, machen sich die Forscher an ein großes Puzzlespiel: Sie nehmen dafür zunächst eine größere Zahl an Zellen, zerteilen dann deren Erbgut in viele kleine Schnipsel – sogenannte „Reads“ – und sequenzieren die Information, die auf diesen kleinen Schnipseln steht. Dies ist notwendig, da die heutigen Techniken nur kleine DNA-Abschnitte verarbeiten können.
Heraus kommt eine riesige Menge an Daten – Milliarden von Reads, ein Datenvolumen von mehreren hundert Gigabyte. Sie bestehen aus unterschiedlich langen Sequenzen aus den Buchstaben A, C, G und T. Die Aufgabe von Bioinformatikern ist nun, deren Position innerhalb eines Chromosoms zu bestimmen, dann die entstehenden Abschnitte einem Chromosom (das sogenannte „Mapping“) zuzuordnen und schließlich noch den richtigen Kopien des Chromosoms zu finden. Letzteres nennt man „Phasing“. Erschwert wird die Aufgabe durch Sequenzierungsfehler, wodurch eigentlich gleiche Teile unterschiedliche Buchstabenkombinationen aufweisen können.
Für das Mapping gibt es gute und effiziente Tools. Noch unzureichend sind die bioinformatischen Werkzeuge für das Phasing. Genau darauf hat sich ein Team von Bioinformatikern der HHU konzentriert. In einem gemeinsamen, DFG-geförderten Projekt unter Leitung von Prof. Dr. Gunnar Klau (Arbeitsgruppe Algorithmische Bioinformatik) und Prof. Dr. Tobias Marschall (Institut für Medizinische Biometrie und Bioinformatik, Universitätsklinikum Düsseldorf) und in Zusammenarbeit mit Prof. Dr. Björn Usadel (Institut für Biological Data Science) haben sie das Softwaretool „WhatsHap polyphase“ entwickelt und erfolgreich sowohl an Modelldaten als auch am Genom der Kartoffel getestet.
Das neue Tool löst das Problem in einem zweiphasigen Prozess. Zunächst werden die Reads geclustert, also in Gruppen aufgeteilt. Reads in einer Gruppe kommen wahrscheinlich von einem Haplotypen oder aus einer Region identischer Haplotypen. In einer zweiten Phase werden die Haplotypen durch die Cluster „gefädelt“. Hierbei werden die Reads möglichst gleichmäßig auf die Haplotypen verteilt und es wird darauf geachtet, dass diese möglichst wenig zwischen Clustern hin- und herspringen.
Das neue Tool wurde in das übergeordnete, frei verfügbare Paket „WhatsHap“ eingespielt. Dieses war bisher in der Lage, erfolgreich das Phasing bei diploiden Chromosomensätzen wie dem des Menschen durchzuführen. Mit der neuen Ergänzung des Düsseldorfer Teams ist nun auch das Phasing bei polyploiden Organismen möglich. Dazu Prof. Klau: „Mit unserer neuen Technik kann nun das Erbgut von Pflanzen in hoher Auflösung und mit geringer Fehlerrate gephased werden.“
Originalveröffentlichung
Weitere News aus dem Ressort Wissenschaft


















Diese Produkte könnten Sie interessieren

Limsophy LIMS von AAC Infotray
Optimieren Sie Ihre Laborprozesse mit Limsophy LIMS
Nahtlose Integration und Prozessoptimierung in der Labordatenverwaltung

OMNIS von Metrohm
OMNIS – die Plattform zur Integration der Metrohm Titrando Gerätegeneration
OMNIS ermöglicht die Kombination von Bestandskomponenten mit neuester OMNIS Hard- und Software

LAUDA.LIVE von LAUDA
LAUDA.LIVE - Die digitale Plattform für Ihre Geräteverwaltung
Viefältige Flottenmanagementoptionen für jedes LAUDA Gerät - mit und ohne IoT-Anbindung





































