Researchers investigate structural changes in snap-frozen proteins
A novel method allows visualization of molecular motions lasting a few millionths of a second
Researchers at the University of Bonn and the research center caesar have succeeded in ultra-fast freezing proteins after a precisely defined period of time. They were able to follow structural changes on the microsecond time scale and with sub-nanometer precision. Owing to its high spatial and temporal resolution, the method allows tracking rapid structural changes in enzymes and nucleic acids.
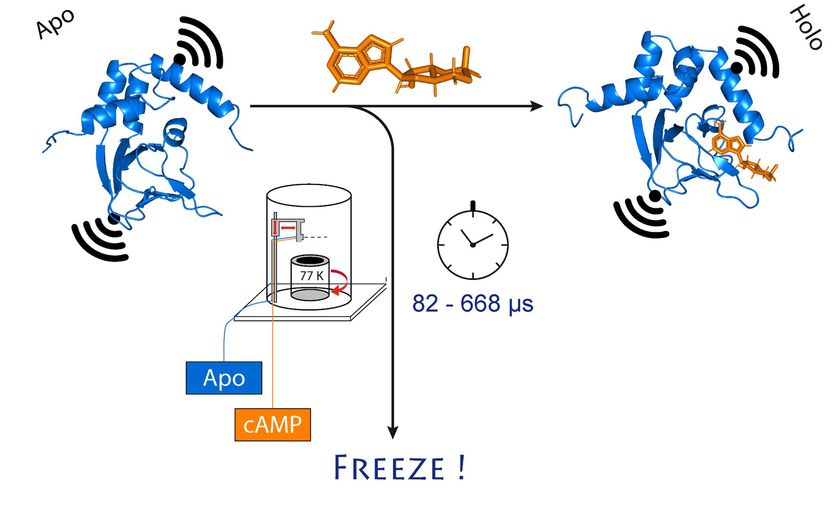
A subunit (blue) of the potassium ion channel changes its arrangement - when cAMP (orange) binds. This also changes the distance between the two molecular magnets attached to the channel (black).
© Tobias Hett / University of Bonn and research center caesar
If you want to know what the spatial structure of a biomolecule looks like, you have a formidable arsenal of tools at your disposal. The most popular ones are electron microscopy and X-ray diffraction, which can reveal even the smallest details of a protein. However, a significant limitation of those methods is that they usually deliver static images, which are often insufficient to understand biomolecular processes in precise mechanistic terms. Therefore, a long-term goal of many research groups worldwide has been to track the movements within a macromolecule such as a protein over time while it carries out its work, just like in a movie. The research groups led by Prof. Dr. Olav Schiemann from the Institute of Physical and Theoretical Chemistry at the University of Bonn and Prof. Dr. Benjamin Kaupp from the research center caesar of the Max Planck Society have now come a step closer to achieving this goal.
They chose an ion channel for their investigation. This is a protein that forms miniscule pores in the cell membrane that are permeable to charged particles called ions. "This channel is normally closed," Schiemann explains. "It only opens when a cellular messenger, called cAMP, binds to it. We wanted to know how exactly this process works."
Mini magnets to measure distances
To do so, the researchers first mixed the channel protein and cAMP and then rapidly froze the solution. In the frozen state, the protein structure can now be analyzed. For their method to work, they had attached molecular electromagnets at two points in the channel. The distance between these magnets can be determined with a precision of a few Angstrom (ten billionths of a millimeter) using a sophisticated method called PELDOR, which works like a molecular ruler. In recent years, the method was significantly refined and improved in Schiemann's group.
"However, this only gives us a static image of cAMP binding to the ion channel," Schiemann says. “We therefore repeated the freezing process at different times after mixing the two molecules. This allowed reconstructing the movements in the protein after cAMP binding - just like a movie, which is also made up of a sequence of images."
At the center of this procedure is a sophisticated method that allows samples to be mixed and frozen very quickly at a precise point in time. The technique, called "microsecond freeze hyperquenching" (abbreviated MHQ), was originally developed at Delft University, but later fell into disuse. It was rediscovered and decisively refined by Kaupp's group.
"In the MHQ device, the cAMP molecule and the ion channel are mixed at ultrafast speed," Kaupp explains. "Then the mixture is shot as a hair-thin stream onto a very cold metal cylinder at –190 °C, which rotates 7,000 times per minute. It was particularly challenging to transfer the frozen samples for the PELDOR measurement from the metal plate into thin glass tubes, and to keep them frozen meanwhile. We had to design and build special tools for that."
Deep-freezing in 82 millionths of a second
The entire mixing and freezing process takes only 82 microseconds (one microsecond equals a millionth of a second). "This allows us to visualize very rapid changes in the spatial structure of proteins," explains Tobias Hett, one of the two doctoral students who contributed significantly to the success. The advantage of the method is its combination of high spatial and temporal resolution. "This represents a major step forward in studying dynamic processes in biomolecules," Kaupp emphasizes.
The researchers now plan to use their method to take a closer look at other biomolecules. They hope to gain new insights, for example into the functioning of enzymes and nucleic acids. The importance of such insights is best illustrated by the recent worldwide surge of structural research on the SARS coronavirus-2: The so-called spike protein of the virus also undergoes a structural change when human cells are infected. Clarifying this mechanism will provide valuable information how to target the infection mechanism with new drugs.
The preparation of the samples, the experimental execution, and the analysis of the data is very complex. The results of the study therefore also reflect a successful scientific cooperation with researchers led by Prof. Dr. Helmut Grubmüller of the Max Planck Institute for Biophysical Chemistry in Göttingen and Prof. Dr. Heinz-Jürgen Steinhoff of the University of Osnabrück.
Original publication

Other news from the department science




















These products might interest you

Kjel- / Dist Line by Büchi
Kjel- and Dist Line - steam distillation and Kjeldahl applications
Maximum accuracy and performance for your steam distillation and Kjeldahl applications

AZURA Purifier + LH 2.1 by KNAUER
Preparative Liquid Chromatography - New platform for more throughput
Save time and improve reproducibility during purification

Most read news






More news from our other portals






























See the theme worlds for related content
Topic world Protein analytics
Protein analytics provides a deep insight into these complex macromolecules, their structure, function and interactions. It is essential for discovering and developing biopharmaceuticals, understanding disease mechanisms, and identifying therapeutic targets. Techniques such as mass spectrometry, Western blot and immunoassays allow researchers to characterize proteins at the molecular level, determine their concentration and identify possible modifications.

Topic world Protein analytics
Protein analytics provides a deep insight into these complex macromolecules, their structure, function and interactions. It is essential for discovering and developing biopharmaceuticals, understanding disease mechanisms, and identifying therapeutic targets. Techniques such as mass spectrometry, Western blot and immunoassays allow researchers to characterize proteins at the molecular level, determine their concentration and identify possible modifications.